In this study, we demonstrate that plasmids are prevalent across both slowly and rapidly growing NTM species, including human pathogens and clinically irrelevant strains. However, the presence of these plasmids is inconsistent across species, subspecies, and even within (sub)lineages, with some strains carrying a specific plasmid while others do not. NTM plasmids are highly diverse with many uncharacterized genes and only a limited number of known resistance or virulence-associated genes. Closely related plasmids were frequently found in different NTM species, suggesting that plasmid-mediated horizontal gene transfer may play an important role in NTM evolution.
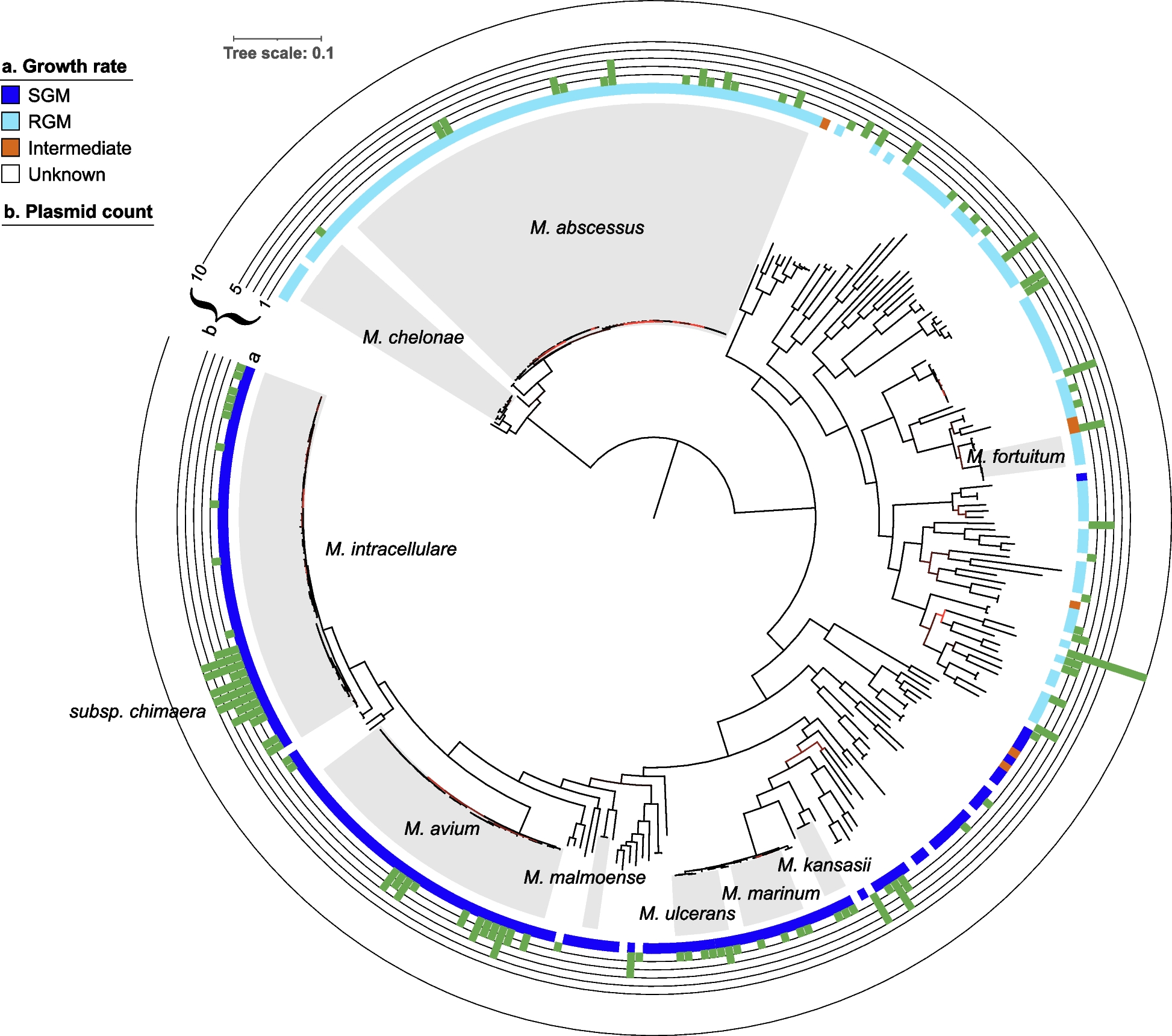
Sequences annotated as plasmids were present in about 30% of all NTM species for which complete genomes were available in NCBI, including clinically relevant species such as M. abscessus, M. avium, M. intracellulare, M. kansasii, M. marinum, and M. ulcerans. However, with only a limited number of complete genomes currently available for many NTM species, more plasmids are expected to be discovered as additional sequencing data becomes available. The majority of NTM genomes containing multiple plasmids harbored between two and five plasmids. The genome of Mycobacterium sp. SMC-4, however, included a closed circular chromosomal sequence and an unusually high number of 10 sequences annotated as plasmids (eight of which were linear and three ≤ 5 kbp) suggesting these contigs may not represent fully assembled functional plasmid entities.
Within NCBI, actually, a large number of NTM plasmids are labeled as linear (17%). Linear NTM plasmids with invertron-like structures (i.e., with terminal inverted repeats) and lengths between 15 and 320 kbp have been described for several NTM including M. xenopi, M. branderi, M. intracellulare, M. celatum, M. abscessus, and M. avium [54,55,56,57,58,59]. Their topology was confirmed by PFGE migration patterns, sensitivity to exonuclease III (which degrades DNA from free 3′ ends), sensitivity to exonuclease lambda (which degrades DNA from free 5′ ends), topoisomerase (which relaxes circular plasmids, changing migration speed) insensitivity and/or RFLP analysis. TIRs were not identified in the SMC-4 sequences nor in most other supposed linear plasmid sequences; however, for some, the ends of the sequences closely matched their beginnings, though not perfectly, suggesting they may be circular but were likely affected by assembly challenges. In addition, we also observed several inconsistencies in submitted annotations. For example, pMyong2, a plasmid from M. intracellulare was experimentally verified to be linear [60], but labeled in NCBI as circular. On the other hand, pMUM002 from M. liflandii [61] was identified as circular by sequencing of overlapping BAC clones while it is labeled as linear in NCBI. In addition, 10 out of 31 clusters of closely related plasmids, comprised plasmids with different topologies (e.g., cluster 28, comprising pMUM001 plasmid) and different plasmid lengths further indicating potential mislabeling or assembly challenges.
Interestingly, not all strains belonging to the same phylogenetic group harbored the same number or type of plasmids indicating several independent events of plasmid acquisition and loss. Notably, nearly all M. intracellulare subsp. chimaera isolates harbored multiple plasmids but even within this subspecies, different plasmid presence patterns were observed in different sublineages. In addition, known plasmids were absent in the genomes of M. avium subsp. avium, the etiological agent of avian tuberculosis, and M. avium subsp. paratuberculosis, a globally important obligate pathogen of domestic and wild ruminants and the causative agent of Johne’s disease [30, 62], while M. avium subsp. hominissuis, which is typically isolated from humans [30], harbored between 0 and 4 plasmids, potentially reflecting differences in their ecology, pathogenicity, host specificity, plasmid uptake potential, or adaptation strategies.
The absence of a protein family universally present across all annotated NTM plasmids suggests either significant diversity in plasmid backbones, potential inaccuracies in sequence annotation, or a combination of both. Indeed, some sequences labeled as plasmids in NCBI may in fact represent misannotated chromosomal fragments, genomic islands, or other mobile genetic elements. However, the in silico validation of the analyzed sequences as true plasmids remains difficult for two reasons. First, many in silico plasmid prediction tools rely on, or are trained with, plasmid data derived from NCBI, i.e., the same data used in this study. Any incorrectly annotated sequence in NCBI can therefore also introduce biases into these prediction tools, resulting in the classification of non-plasmid sequences as plasmids. On the other hand, current plasmid prediction tools may fail to identify true plasmid sequences due to reliance on outdated databases, overly strict filtering criteria (e.g., plasmid length thresholds unsuitable for NTM plasmids), or an inability to detect highly divergent plasmid backbone genes (e.g., replication genes). This might be particularly problematic for NTM plasmids, as there is a lack of experimental verification to confirm their existence, replication mechanisms, and other key features.
Genes most prevalent on the presumed NTM plasmids were either annotated as hypothetical proteins or mostly related to basic plasmid functions such as replication (e.g., repA), maintenance of plasmid copy number and evolution (e.g., recombinases/integrases), segregation (e.g., toxin/anti-toxin systems), mobilization (e.g., mob relaxases) and conjugation (e.g., type VII secretion system). Putative resistance genes were identified in both human-pathogenic and non-pathogenic mycobacterial plasmids, though only a few are predicted to confer resistance to the antibiotics most commonly used to treat NTM infections: aminoglycosides, macrolides, and rifamycin. Additionally, amino acid identity compared to resistance proteins in the AMRfinder + reference database was typically low (< 55%), underscoring the need for in vitro (e.g., phenotypic drug susceptibility testing) and in vivo (e.g., using antibiotic-treated infected mice) experiments to confirm their function in NTM. This is further highlighted by the fact that the notorious IncP1 multi-drug plasmid (NC_017908.2, BRA100) from the M. abscessus subsp. massiliense strain that caused the nation-wide post-surgical infection outbreak in Brasil [9, 10] contains three genes that encode putative resistance against aminoglycoside antibiotics. However, all investigated epidemic Brazilian strains so far showed phenotypic susceptibility against amikacin [29,30,31,32]. Genes for known beta-lactamases were solely found on the chromosomes and not on NTM plasmids. The erm(55)P gene, recently identified as potentially conferring plasmid-mediated inducible macrolide resistance in M. chelonae, was found in our dataset solely on the original plasmid pMchErm55 [63, 64] and in one M. obuense draft genome. Nonetheless, continued surveillance of both the gene and plasmid is advised.
The only putative virulence sequence that was detected with AMRfinder + relaxed parameter settings was yfeB, found on one plasmid belonging to the pathogen M. ulcerans and on two plasmids from M. aubagnense, which rarely causes disease in humans. This gene, coding for an iron/manganese ABC transporter ATP-binding protein, was described in the plague pathogen Yersinia pestis, where it was shown to play an important role in iron acquisition and virulence [65]. However, given that the putative yfeB sequences encoded on the NTM plasmids share only 31% amino acid identity with those from Yersinia, there is a fair chance that these proteins may not perform the same role. Additional homologs of M. tuberculosis virulence factors [47] that were not detected by AMRfinder + were found in almost 25% of NTM plasmids but at least some of them might also be pseudogenes. Plasmids closely related to the supposed virulence plasmid pMAH135 [13] were only found in members of the MAC.
Clustering or classifying plasmids is crucial for understanding their genetic diversity, evolutionary relationships, and functional roles, as well as for tracking the spread of antibiotic resistance and virulence factors. Until now, plasmid classification and characterization efforts have mainly been focused on plasmids from Enterobacteriaceae [40, 66]. Based on sequence similarity of their replication genes and the inability to coexist in the same cell, Enterobacteriaceae plasmids have been classified into so-called incompatibility (inc) groups [67]. In addition, inc groups can be subtyped using plasmid multi-locus sequence typing (pMLST) [40], and transmissible plasmids can be classified based on relaxase genes (MOB-typing). Identified mobility proteins from NTM plasmids all belonged to two out of six known MOB families [68], i.e., mobF and mobP. However, only the plasmid from the M. abscessus clone BRA-100 could be assigned to an existing incompatibility group, demonstrating that this typing method has limited applicability for NTM.
Therefore, we clustered the annotated NTM plasmids based on their overall sequence similarity to one another and screened for their presence in draft assemblies and short-read sequencing data from thousands of NTM isolates. We observed that many closely related plasmids were shared among multiple NTM species, including some distantly related species, while others were restricted to specific phylogenetic groups within a species. This suggests that both horizontal gene transfer and vertical inheritance are likely mechanisms of plasmid acquisition within NTM. Horizontal gene transfer across species might be facilitated by the fact that different NTM species can occupy the same environmental niche [69, 70] and can also co-colonize or infect patients simultaneously [25, 71,72,73]. On the other hand, some detections of plasmids in NTM species other than the original host may also be due to low-level undetected contamination, potentially from cultures that were not thoroughly subcultured to achieve pure isolates.
A limitation of this study is its reliance solely on in silico data. Although all 196 analyzed putative plasmid sequences were derived from complete genomes, labeled as plasmids in the NCBI database, and included in the curated plasmid database PLSDB [41], we cannot rule out the possibility that some sequences, apart from those of SMC-4, may be incomplete or incorrectly classified as plasmids. In addition, we did not specifically search for novel plasmids, as reconstructing complete plasmid sequences from short-read data remains extremely challenging [74], particularly for NTM genomes, which often harbor multiple large plasmids. Still, even long-read sequencing techniques like PacBio and Nanopore, which are typically well-suited for fully assembling plasmid sequences, seem to struggle with this, at least for some NTM isolates. Additionally, it must also be noted that plasmids may have been lost during subculturing, DNA extraction, or library preparation prior to sequencing biasing prevalence numbers. Lastly, as with plasmid backbone genes, it is also possible that putative resistance and virulence genes located on NTM plasmids are overlooked, i.e., not detected with current in silico prediction tools, because they are not yet well-characterized or not included in current reference databases. To address this, we also applied more relaxed detection thresholds, though this comes with the drawback of potentially increasing false positive results [45].







Comments (0)