
Remember me

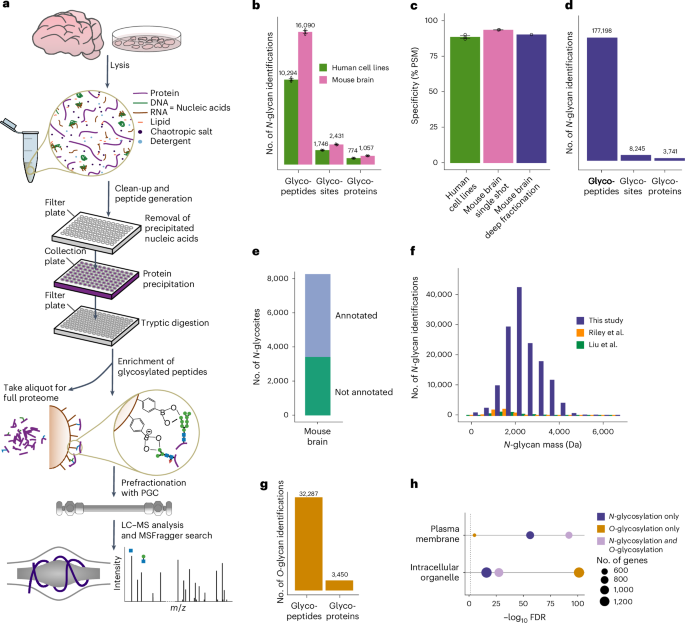

We developed a glycoproteomics workflow (Fig. 1a) using commercially available, economical silica beads functionalized with PBA to selectively enrich intact glycopeptides. All steps of the workflow, from sample preparation to glycopeptide enrichment, were performed in 96-well (filter) plates, increasing sample processing throughput. In addition to glycopeptides, other diol-containing biomolecules, such as RNA, can also bind to PBA beads, interfering with the detection of glycopeptides. This is illustrated by the high abundance of an RNA marker ion (330.06 m/z)11 in glycopeptide-enriched samples after 2% strong denaturing detergent (SDS) lysis followed by protein precipitation (Supplementary Fig. 1a). To address this, we optimized the sample lysis buffer by incorporating a high concentration of chaotropic salts and organic solvent to induce nucleic acid precipitation while proteins remained in solution. Nucleic acid aggregates were then filtered out on 96-well filter plates, followed by protein precipitation by further increasing organic solvent concentration before tryptic enzymatic digestion. This fast sample preparation workflow (1 h for 96 samples) enabled efficient removal of RNA molecules (Supplementary Fig. 1a), increasing the number of unique N-glycopeptides identified by 60% (Supplementary Fig. 1b and Supplementary Data 1).
Fig. 1: Deep profiling of protein glycosylation in human cell lines and mouse brain.
a, DQGlyco workflow. b, Average numbers of unique glycopeptides, glycosites and glycoproteins identified from LC–MS/MS triplicates of unfractionated samples from human cell lines (HelaK and HEK293T cells) and mouse brain. Data are presented as the mean values ± s.d. c, Glycopeptide enrichment specificity, calculated as the ratio of glycopeptide spectrum matches to all peptide spectrum matches (PSM) for unfractionated samples from human cell lines (HeLaK and HEK293T; n = 3 technical replicates per cell line) and mouse brain (n = 3 technical replicates), as well as from PGC-fractionated mouse brain samples. Data are presented as the mean values ± s.d. d, Total number of identified N-glycopeptides (unique sequence and glycan composition), N-glycosites and N-glycoproteins from PGC-fractionated mouse brain samples. e, UniProt and PhosphoSitePlus annotation of N-glycosites identified in mouse brain samples. f, Total number of unique glycopeptides of the PGC-fractionated mouse brain sample per binned m/z range compared to recent large-scale glycoproteomics studies (Riley et al.16 and Liu et al.17). g, Total number of O-glycopeptides and O-glycoproteins identified in the PGC-fractionated mouse brain sample. h, GO enrichment analysis (STRINGdb) for proteins found to be only N-glycosylated, only O-glycosylated or both N-glycosated and O-glycosylated for two selected terms (intracellular organelle and plasma membrane).
Next, we adjusted the full scan (first-level mass spectrometry (MS1)) range to preferentially target glycopeptides that are higher in mass compared to nonmodified peptides that nonspecifically bind to PBA beads (Supplementary Fig. 1c). This increased the number of identified unique N-glycopeptides by 18% and the enrichment specificity of N-glycopeptides by 13% compared to a commonly used mass over charge scan range (Supplementary Fig. 1d,e and Supplementary Data 1). In total, using MSFragger12 as a search engine, we were able to identify an average of 10,294 unique glycopeptides, 1,746 glycosites and 774 glycoproteins in human cell lines (HeLa and HEK293T; 1% false discovery rate (FDR) at the N-glycopeptide level, glycan q values ≤ 0.05) per single-shot replicate. These numbers increased to 16,090 unique glycopeptides, 2,431 glycosites and 1,057 glycoproteins in mouse brain samples (Fig. 1b). Overall, deep quantitative glycoprofiling (DQGlyco) enables high-throughput sample preparation and enrichment of hundreds of samples per day. Moreover, DQGlyco markedly improves the detected glycoproteome coverage when compared to previous studies (Supplementary Fig. 1f) without the need for prefractionation. Remarkably, the enrichment selectivity exceeded 90% for all samples (Fig. 1c and Supplementary Data 1).
Deep mouse brain glycoproteome analysisTo increase the glycoproteome coverage in mouse brain samples, we used porous graphitic carbon (PGC) as a first dimension of chromatographic separation for glycopeptides before a second online C18-based reversed-phase separation. Leveraging a mixed-mode retention mechanism13, PGC can more efficiently resolve different glycan species compared to traditional reversed-phase chromatography14,15; thus, as a first dimension of separation, PGC should facilitate a better capture of glycosylation microheterogeneity. Our approach identified 177,198 unique N-glycopeptides and 8,245 N-glycosites located on 3,741 N-glycoproteins (Fig. 1d and Supplementary Data 2). This represents a greater than 25-fold improvement in the identification of unique N-glycopeptides compared to current state-of-the-art N-glycoproteomics studies on mouse brain samples16,17 (Supplementary Fig. 1f). Notably, this improvement persists when searching previous datasets with our analytical pipeline (Supplementary Fig. 1g) or searching one of our datasets with another search engine (Byonic18; Supplementary Fig. 1h).
The majority of identified glycoforms were on annotated plasma membrane or extracellular proteins (Supplementary Fig. 1i). No differences in glycopeptide molecular weight and peptide length were observed between the single-shot and fractionated samples, indicating that PGC fractionation does not introduce a bias for the detection of shorter, more readily eluted glycopeptides (Supplementary Fig. 1j).
Our dataset encompasses 65% of the proteins that have been previously identified in various regions of the mouse brain19 and annotated as glycosylated in UniProt (Supplementary Fig. 2a). Overall, around half of the glycosites and glycoproteins identified in this study were reported as glycosylated in the UniProt and PhosphoSitePlus databases, with only 6% having experimental validation evidence (Fig. 1e and Supplementary Fig. 2b). Thus, this study substantially expands the known mouse brain N-glycoproteome. Proteins reported as glycosylated in UniProt are predominantly annotated as part of the plasma membrane while previously unreported glycosylated proteins are mainly associated with subcellular organelles (Supplementary Fig. 2c). The identification of 332 glycoproteins in our dataset that were not found in the mouse proteome atlas19 study (Supplementary Fig. 2d) highlights the sensitivity of our method as these proteins are likely expressed at low levels. Typically, glycopeptide enrichment methods exhibit biases toward certain glycan compositions. We compared the different glycan compositions or masses identified by alternative enrichment strategies and found that N-glycans identified by our PBA-based approach encompass those previously identified by different approaches (Fig. 1f and Supplementary Fig. 2e) while greatly improving the glycoproteome coverage, suggesting a low enrichment bias toward specific glycopeptides.
Understanding the stoichiometry of different glycoforms is important for interpreting the biological implications of glycoform alterations. In this study, we can only perform relative quantification as glycan composition can impact glycopeptide ionization efficiency (Supplementary Fig. 2f). Using MS1 intensities as a proxy to calculate glycoform fractional intensity (ratio of glycoform intensity to the sum of all glycoform intensities at a given site) revealed that most glycoforms exhibit a low fractional intensity (Supplementary Fig. 2g). When focusing on the glycoform having the highest fractional intensity per site (here, only sites with at least ten glycoforms were considered, representing 1,580 sites), it becomes clear that glycosylation of a specific site is rarely dominated by a single glycoform of high stoichiometry (Supplementary Fig. 2h).
N-glycopeptides are more abundant than O-glycopeptides in mammalian cells; hence, O-glycopeptide identification usually necessitates dedicated analytical strategies7. We reasoned that the deep glycoproteome coverage obtained by DQGlyco could enable the simultaneous identification of both N-glycopeptides and O-glycopeptides. In total, we identified 32,287 O-glycopeptides on 3,450 O-glycoproteins in mouse brain samples (Fig. 1g and Supplementary Data 2), constituting an extensive characterization of intact O-glycosylation in a mammalian tissue. It has to be noted that, as higher-energy collisional dissociation (HCD) fragmentation was used in this study, we could not confidently assign the site of glycosylation within a given glycopeptide20. We could identify over 494 unique O-glycan compositions, with a molecular weight distribution shifted to lower masses when compared to N-glycans (Supplementary Fig. 2i). A total of 1,853 proteins were only O-glycosylated and not N-glycosylated. Proteins that were solely O-glycosylated were enriched in intracellular membrane-bound organelles (Fig. 1h). In accordance with previous reports, O-linked N-acetylhexosamine (O-HexNAc) modification was enriched in intracellular organelles, whereas no difference in glycan elongation was observed between proteins belonging to the plasma membrane and to intracellular organelles (Supplementary Fig. 2i). In addition, using oxonium ion ratios for analysis21, we observed that O-glycans on proteins annotated as intracellular were predominantly modified by N-acetylglucosamine (GlcNAc), while O-glycans on proteins annotated as extracellular or belonging to the plasma membrane were predominantly modified by N-acetylgalactosamine (GalNAc)22 (Supplementary Fig. 2j). Conversely, N-glycans were predominantly modified by GlcNAc, regardless of the protein subcellular localization (Supplementary Fig. 2k).
DQGlyco reveals extensive microheterogeneityAlthough half of the sites identified were modified by one or two glycoforms, numerous sites exhibited high microheterogeneity, with an average of 17.4 glycoforms per site (Fig. 2a) and extreme cases such as the N205 site on the excitatory amino acid transporter 2 protein (Eaat2, also referred to as Slc1a2), on which 667 glycoforms were identified. No correlation was observed between the detected number of glycosites per protein or number of glycoforms per site and the abundance of the respective protein (Supplementary Fig. 3a,b). Similarly, no major differences in isoelectric point and peptide length were observed for glycopeptides bearing sites of different microheterogeneity (Supplementary Fig. 3c,d). This suggests that the glycosylation heterogeneity observed is not skewed toward easily detected or abundant glycoproteins and glycopeptides.
Fig. 2: Protein glycosylation composition and microheterogeneity in the mouse brain.
a, Frequency of the number of unique glycan compositions per site in the PGC-fractionated mouse brain N-glycosylation data (on average, 18 glycan compositions were identified per site). b, Total number of unique glycopeptides, glycosites and glycoproteins per glycan class for the PGC-fractionated mouse brain sample. c, Frequencies of glycan classes identified in this study and in a recent glycoproteomics study using lectin-based enrichment16. d, Distinct glycan classes are significantly overrepresented or underrepresented on specific functional protein domains from the InterPro database (log2 odds ratio > 0 or log2 odds ratio < 0, Fisher’s exact test, adjusted P value < 1 × 10−3; examples shown). e, Glycosylation profiles of sites belonging to the same protein are significantly more correlated compared to when glycan compositions are shuffled across the whole glycoproteome (Kendall rank correlation coefficient for n = 29,932 site pairs per distribution, two-sided Wilcoxon rank-sum test, P < 2.2 × 10−16). Box plots indicate the median and the first and third quartiles. Whiskers extend from the hinges to the largest value no further than 1.5 × the interquartile range. Data points beyond the end of the whiskers are plotted individually. f, Results of the GO enrichment analysis (STRINGdb) of proteins with sites displaying low, medium or high microheterogeneity (n = 1–2, 3–11 and 11+ unique glycan compositions per site, respectively) for selected significant terms (two-sided Fisher’s exact test, FDR < 0.05). g, Frequency of glycosites or glycopeptides with a close known phosphosite (±5 residues) per glycan class for O-glycosylation and N-glycosylation data.
The different glycan compositions were grouped into eight classes, which can have varying impacts on protein–protein or protein–ligand interactions through steric or electrostatic effects and can serve as epitopes for specific lectin receptors1,2,3,4. N-glycans are added in the ER as high mannose and can be processed in the Golgi into glycans classified as high mannose, small, paucimannose, phosphorylated, fucosylated and sialylated, while the remaining compositions were considered as complex or hybrid (Fig. 2b and Supplementary Fig. 3e,f; details in Methods). Compared to the Riley et al. dataset16, we identified markedly less high-mannose N-glycosylation (fewer than 15% of all glycopeptides identified versus 60%). This confirms that the commonly used conA lectin-based enrichment is biased toward high-mannose glycosylation and underscores the need for deep analytical coverage to achieve comprehensive characterization of mature glycoforms (Fig. 2c). In this study, close to half of the sites modified by only one glycoform were found to be modified by high mannose, implying that these sites are indeed subjected to a low number of processing events (Supplementary Fig. 3g).
The distribution of different classes of N-glycosylation differs between the two human cell lines and the mouse brain (Supplementary Fig. 3h), likely reflecting differences in the regulation of glycosylation processes. Despite the fact that little difference in enriched Gene Ontology (GO) terms was observed between the different glycan classes (Supplementary Fig. 3i), meaning that different types of glycosylation in general do not target proteins with specific functions, significant distinctions were observed in the enriched protein domains for the different N-glycosylation classes (Fig. 2d). Na+/K+-transporting adenosine triphosphatase domains were, for example, strongly enriched in sialylated and paucimannose glycans, while saposin domains (specific to lysosomal proteins) were enriched for phosphoglycans. In general, phosphoglycans showed strong enrichment on lysosomal proteins (Supplementary Fig. 3j), aligning with the role of mannose-6-phosphate as a tag directing enzymes to the lysosome23.
As the different sites of N-glycoproteins should be exposed to the same glycoenzymes, we investigated if microheterogeneity across different sites within the same protein was more similar than across the glycoproteome. We observed no clear pattern of co-occurrence of different classes of N-glycosylation either at the glycosite or at the glycoprotein level (Supplementary Fig. 4a,b). However, when correlating the exact N-glycoform compositions between sites localized on the same protein before and after random reshuffling of all glycoforms across all identified sites, we concluded that, while there is substantial variation in microheterogeneity between different sites on the same protein, this variation is significantly lower than across the glycoproteome as a whole (Fig. 2e).
Differences were observed when classifying N-glycoproteins according to the level of site microheterogeneity, independently of glycan compositions; sites with high heterogeneity were enriched on extracellular proteins, involved in processes such as cell-surface receptor signaling pathways and cell adhesion, both known to be modulated by glycosylation2 (Fig. 2f). Interestingly, the level of site microheterogeneity within a given glycoprotein tended to be site specific (Supplementary Fig. 4c).
No specific protein annotation was associated with a class of O-glycosylation, with intracellular or plasma membrane O-glycoproteins exhibiting relatively similar glycosylation patterns (Supplementary Fig. 4d). However, O-glycopeptides on which only one glycoform was identified were predominantly modified by a single HexNAc sugar (Supplementary Fig. 4e) and O-HexNAc modification occurred mostly on intracellular proteins (Supplementary Fig. 4f). Similarly, proteins with only one identified O-glycoform were enriched in intracellular organelle proteins while proteins with higher heterogeneity were enriched in membrane proteins involved in signaling (Supplementary Fig. 4g). It has been reported that O-GlcNAcylation (addition of a single sugar) and phosphorylation have a propensity to co-occur on the same protein regions, resulting in posttranslational modification (PTM) crosstalk24. Here, we show that around 25% of O-glycosylated peptides were also previously reported to be phosphorylated, independently of the glycan mass and composition, suggesting that the existence of crosstalk can be extended to all glycoforms on O-glycosites. In comparison, fewer than 2% of N-glycosites were in close proximity to reported phosphosites (Fig. 2g).
Machine learning prediction of site microheterogeneityUsing AlphaFold predictions25, we extracted structural variables for glycosylated asparagine residues, including prediction-aware part-sphere exposure (pPSE), which calculates how many amino acid residues are in the structural vicinity of each site while accounting for AlphaFold prediction error. Additionally, AlphaFold predictions were used to annotate each glycosite with its predicted secondary structure25,26. We used this information to define intrinsically disordered regions (IDRs), while topological domain annotations were retrieved from UniProt. Using glycan classes to group the sites in different categories, we found little differences between sites, except for those where we only detected high-mannose glycans, which tend to be located in less structured regions (Supplementary Fig. 5a). We then investigated the variability of structural features after grouping sites on the basis of their level of microheterogeneity, revealing that sites of high microheterogeneity tended to be located in more structured regions, bends and turns but not in α-helices (Supplementary Fig. 5b). These observations are exemplified on the adenovirus receptor protein CXADR (Fig. 3a,b).
Fig. 3: N-glycosylation in a structural context.
a, Glycosylated asparagines highlighted as spheres in the AlphaFold-predicted structure of the coxsackievirus and adenovirus receptor (P78310). Colors indicate the model predicted local distance difference test (pLDDT). b, Total number of identified glycans and relative glycan type composition per site in the same protein. Topological domains and IDRs are also highlighted (IDR definition in Methods). c, Validation AUROC for the predictive model of lowly and highly glycosylated asparagines that uses structural features as input (details in Methods; low-N = 2,203 and high-N = 747 in mouse; low-N = 1,196 and high-N = 488 in human). d, Feature importance plot. The x axis indicates the F score for every feature on the y axis. e, pPSE (24 Å, 180°) for highly and lowly glycosylated asparagines (low-N = 2,203 and high-N = 747 in mouse; low-N = 1,196 and high-N = 488 in human). f, Log-transformed number of glycoforms on asparagines within extracellular (N = 2,813) or cytoplasmic (N = 335) topological domains. g, Average AlphaMissense score for lowly (N = 1,196) and highly (N = 488) glycosylated asparagines on data from human cell lines. All box plots indicate the median and the first and third quartiles. Whiskers extend from the hinges to the largest value no further than 1.5 × the interquartile range. Data points beyond the end of the whiskers are plotted individually. All P values indicate the comparison of distributions using a two-sided Wilcoxon rank-sum test.
Using these variables as input, we built a model to discriminate between sites of low and high microheterogeneity (Methods). Assuming that a structural signal would be conserved between mouse and human, we fitted our model on sites identified in the mouse brain glycoproteome and validated it using data from human cell lines. This model achieved a validation area under the receiver operating characteristic curve (AUROC) of 0.707 (Fig. 3c). The feature importance analysis revealed that pPSE (24 Å, 180°) and pPSE (12 Å, 70°) were the most relevant features for the predictive power of the model (Fig. 3d). As both variables directly inform about the proportion of residues surrounding glycosites, we found that sites of high microheterogeneity tended to be localized in more structured regions compared to sites exhibiting low microheterogeneity (Fig. 3e). Interestingly, when focusing on accessibility at non-IDRs, we found the inverse pattern, with sites of high microheterogeneity being more exposed (Supplementary Fig. 5c). This suggests that specific ordered structures combined with high site solvent accessibility are needed for extensive glycan processing by glycosyltransferases.
As expected, we found that extracellular glycosites exhibited a larger extent of glycan microheterogeneity than their cytoplasmic counterparts on the same proteins, in both mice (Fig. 3f) and human (Supplementary Fig. 5d). The above observations are exemplified by the detected glycosylation pattern on the adenovirus receptor protein CXADR (Fig. 3a). Lastly, we leveraged the recently published AlphaMissense model, which predicts the effect of missense variants in the human genome27. We found that asparagine residues exhibiting high microheterogeneity had, on average, a higher AlphaMissense score than low microheterogeneity sites, indicating their relevance for normal protein function and their potential pathogenicity when substituted by other residues (Fig. 3g).
Deep multiplexed quantification of glycoformsTo unravel mechanisms governing glycosylation regulation, robust and high-throughput quantification methods are required. So far, most of the quantitative glycoproteomics studies have relied on label-free approaches, which come with inherent limitations as these strategies are incompatible with the different prefractionation techniques that improve analytical depth. Alternatively, tandem mass tag (TMT) labeling, enabling multiplexing, compatibility with prefractionation and low number of missing values, has been applied in glycoproteomics28,29,30. However, the prohibitive cost of TMT reagents has hindered its routine adoption, as pre-enrichment labeling, where both the glycopeptides and the nonmodified peptides are labeled together, is recommended for accurate quantification. Here, we showcase the feasibility of decreasing the number of TMT reagents used 200-fold through postenrichment labeling, as only the substoichiometric glycopeptides need to be labeled. This cost-effective quantification method allows for the multiplexing of up to 18 conditions with high quantitative reproducibility, as shown in Figs. 4c and 5b and Supplementary Data 4 (average correlation > 0.984 between biological replicates of different mouse tissue samples (liver, kidney and brain) at the glycopeptide level). This underscores the high reproducibility achieved through our sample preparation workflow and enrichment platform. Moreover, this highlights that the glycoproteome is tightly regulated and conserved between biological replicates (see Fig. 7b), in contrast to other tissues such as plasma, for which high interindividual differences were reported31.
Fig. 4: Time course of fucosylation inhibition.
a, Experimental design. b, PCA of the normalized TMT glycopeptide intensities per treatment condition, time point and replicate. PC, principal component. c, Glycopeptide modulation over time. Glycopeptides significantly regulated for at least one time point were grouped in four clusters depending on their kinetics of regulation using neural gas clustering. d, Proportion of glycan classes present in the four clusters. e, Fucosylated peptides being significantly downregulated for at least one time point were grouped into three clusters using neural gas clustering. f, Glycoproteins having fucosylated glycoforms belonging to the three clusters. Here, the mean log2 fold changes of the fucosylated glycopeptides belonging to each cluster are displayed. FC, fold change. g,h, The fucosylated glycoforms of the C-type mannose receptor 2 (MRC2) and the neuronal cell adhesion molecule (NRCAM) exhibit different rates of downregulation.
Fig. 5: Systematic characterization of surface-exposed glycoforms.
a, Intact living HEK293 cells are treated with either PNGase or proteinase K. Surface-exposed glycoforms are affected while intracellular glycoforms remain protected. b, Number of glycoforms on affected sites (at least two glycoforms changing) significantly changing in abundance upon enzymatic treatments, per subcellular compartment annotation of proteins. c,d, Characterization of surface-exposed glycoforms belonging to the zinc ion channel ZIP6 and to the cell adhesion protein DSC2. Each data point represents a unique glycoform, with the color representing the glycan class. The shape of the data point reflects significance, while the dotted line represents the effect size cutoff (log2 0.6 = 1.5 fold change). e, Fold change of glycoforms, separated per glycan class, on sites for which at least two glycoforms were affected by the enzymatic treatments. High-mannose glycans were significantly less exposed than the other glycan classes (two-sided t-test). The horizontal lines represent the median. f, Comparison of fold changes of glycopeptide abundance upon proteinase K (y axis) and PNGase treatments (x axis), for sites affected by both enzymes (sites with at least two glycopeptides significantly changing in abundance upon treatment) and glycopeptides identified in both experiments. g, Number of times a given glycan composition was identified on a glycopeptide significantly changing in abundance upon proteinase K (y axis) and PNGase (x axis) treatments. The size of the dot represents the frequency with which this composition was identified as surface exposed.
Fucosylation inhibition reveals glycoform-specific kineticsWe first used our quantitative workflow to study the inhibition of fucosylation over time following treatment with 2-fluorofucose (2FF; Fig. 4a). 2FF treatment leads to a general suppression of protein fucosylation, inhibiting several biological processes such as tumor growth, adhesion and metastasis32. The profiling of the glycoproteome over 3 days of treatment of HEK293f cells revealed a gradual decrease in fucosylation, alongside extensive remodeling across all glycan classes, while no global effect on protein expression was observed (Fig. 4b–d, Supplementary Fig. 6a–c and Supplementary Data 3). Notably, the downregulation of fucosylation occurred at varying rates, which we grouped into three main clusters (Fig. 4e), revealing a widespread site-specific and glycoform-specific modulation. This is illustrated by fucosylated glycoforms of the same protein belonging to different downregulated clusters (Fig. 4f) or the fact that, in general, fucosylated glycoforms at the same site are not coregulated (Supplementary Fig. 6d). This means that such differences in the kinetics of fucosylation downregulation cannot be solely attributed to differences in protein turnover rates. A clear example of this observation is the modulation of glycoforms on the proteins encoded by MRC2 and NRCAM, where fucosylated glycoforms on different sites follow distinct kinetics (Fig. 4g,h).
It is generally accepted that changes in protein glycosylation occur mainly through the modulation of glycoenzyme abundance, localization or activity. In such cases, similar trends would be expected at the glycoproteome level, especially for glycosites within the same protein. This site-specific and glycoform-specific modulation could be explained by glycoform-specific turnover rate or by more complex modulation at the fucosyltransferase level.
Determination of cell-surface-exposed glycoformsN-glycans are added to asparagines as high mannose in the ER before being further processed in the Golgi. Once the cells are lysed, it becomes impossible to differentiate mature, functional glycoforms from intermediate glycoforms going through the secretory pathway. Because of the complexity of the glycosylation machinery, it is not possible to predict the composition of mature glycans and it is postulated that glycoforms of any composition or structure could potentially be surface exposed2. Differences have been observed between cell-surface and intracellular glycans33 but obtaining a comprehensive site-specific overview of mature glycoforms has been challenging so far and the compositions or the proportion of mature glycoforms in a given glycoproteomics experiment remains an open question. We applied our quantitative workflow to intact living human HEK293 cells subjected to either peptide:N-glycosidase F (PNGase F) treatment for 30 min (glycosidase targeting N-
Comments (0)