Dimethylsulfoxide (DMSO) clusters are crucial for understanding chemical processes in DMSO liquid. For example, DMSO clusters can be used to study the solvation of the proton in DMSO [1], [2] or the solvation of any ion in DMSO [3], [4]. Furthermore, DMSO clusters can be used to calculate the proton transfer free energy and enthalpy from DMSO to another liquid or vice-versa [5], [6], [7]. Besides, DMSO clusters can be used along with the quantum cluster equilibrium (QCE) theory to determine the properties of liquid DMSO [8]. The structures of DMSO clusters are essential for the theoretical understanding of most reactions in liquid DMSO. Therefore, it becomes essential to gain enough insights into their geometries, relative and binding energies, their hydrogen bond networks, as well as the effects of temperature on their stabilities. However, the exploration of DMSO clusters’ potential energy surfaces (PESs) to determine the possible structures has received negligible attention.
Venkataramanan and coworkers [9], [10] have studied linear DMSO clusters from dimer to decamer at the B3LYP-D3/6-311++G(d,p) level of theory. The authors have not explored the PESs of the DMSO clusters to locate all possible configurations. The focus was on the linear structures of DMSO, which is the basics of the crystal structure of DMSO [10]. In addition, the binding energies of DMSO clusters have been calculated using several computational levels of theory, including B3LYP, M05-2X, M06-2X, and MP2. The authors provide insights into the hydrogen bondings in the studied linear geometries using the quantum theory of atoms in molecules (QTAIM) analysis [10]. The QTAIM analysis shows that there are two types of non-covalent hydrogen bondings in linear DMSO clusters: “S–O⋯H–C hydrogen bonds and methyl C–H⋯H–C dihydrogen bonds” [10]. Very recently, we performed a thorough exploration of DMSO clusters from dimer to pentamer, starting with molecular dynamics and followed by optimizations at the MP2/aug-cc-pVDZ level of theory [11], [12]. We have been interested in locating all possible structures within a certain energy threshold, contrary to the linear DMSO reported by Venkataramanan and coworkers [9], [10]. After locating all possible structures of the DMSO clusters, we performed a QTAIM analysis of the clusters from dimer to tetramer. It has been found that the CH⋯O hydrogen bondings are the strongest non-covalent interactions, while the H⋯H bondings interactions are the weakest non-covalent bondings in DMSO clusters [11]. Furthermore, a density functional theory (DFT) benchmarking has been performed to identify the most suitable functional to study larger-sized DMSO clusters. It has been found that the functionals PW6B95-D3 and ωB97X-D have the smallest mean absolute deviation from the DLPNO-CCSD(T)/CBS level of theory.
To the best of our knowledge, the studies mentioned above are the only ones reported on the DMSO clusters’ structures in the literature. It comes out from these studies that the investigations of DMSO clusters have been thoroughly performed only from dimer to pentamer. For larger-sized clusters, only the study of linear geometries has been reported by Venkataramanan and coworkers [9], [10]. Therefore, to enable the complete application of DMSO clusters, we thoroughly explored the PESs of the DMSO clusters from dimer to decamer at the PW6B95-D3/def2-TZVP level of theory. As mentioned above, the PW6B95-D3 functional is one of the best functional suitable for studying DMSO clusters. A total of 159 configurations of DMSO clusters have been located from dimer to decamer to calculate the properties of liquid DMSO using the QCE theory.
Quantum cluster equilibrium (QCE) theory extends classical thermodynamics for studying liquids. Weinhold [13] has provided the general theory and its implementation in 1998. Weinhold and his collaborators have applied the QCE theory to study the properties of liquid water [14] and liquid ammonia [15], [16]. Considering the early success of the QCE theory, several authors have adopted it to study the properties of liquids [17], [18], [19], [20], [21], [22], [23], [24], [25]. The group of Kirchner has proposed an extension of the QCE theory to study binary mixtures of liquids (bQCE) [26], [27]. Furthermore, the group has reported several applications of QCE and bQCE [28], [29], [30]. The PEACEMAKER code implementing the QCE and bQCE has been made freely available by the group [8], [31].
Recently, Kuo and coworkers [32] have applied the QCE theory to the study of liquid methanol after thoroughly exploring the PESs of methanol clusters from dimer to dodecamer. The authors have assessed several computational levels of theory [32]. They have provided a Python implementation of the QCE code. In addition, they predicted the population and the infrared spectrum of liquid methanol using four levels of theory. Very recently, we extended the application of the QCE theory to liquid ethanol [33]. We located 484 configurations of the ethanol clusters from dimer to hexamer at the MP2/aug-cc-pVDZ level of theory. The located configurations have been used to determine the population of the liquid ethanol, its infrared spectrum, and some thermodynamic properties of liquid ethanol [33]. Despite the success of the QCE theory, its application has been limited to very few solvents, including water, ammonia, N-methylformamide, N-methylacetamide, methanol, ethanol, and other alcohols. This limitation is because applying QCE requires a thorough exploration of the PESs of the involved clusters. Therefore, an extension of QCE to other liquids after thoroughly exploring the involved molecular clusters will be essential to fill the gap in the literature.
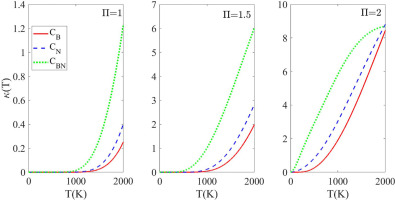
In this work, we used the DMSO clusters’ located structures to extend the QCE application to the liquid DMSO. Therefore, we predicted the population of liquid DMSO and some thermodynamic properties. Besides, we calculated the infrared spectrum of the liquid DMSO using the predicted population. Successful application of the QCE theory requires a thorough exploration of the PESs of the clusters of the solvent molecule. In addition, the cluster size should be as large as possible. However, only clusters with moderate size (less than 12 solvent molecules) usually have meaningful contributions to the liquid population. Furthermore, the accuracy of the QCE theory relies on the accuracy of the total partition function. This implies accurate electronic energies and vibrational frequencies. To satisfy these accuracy requirements, we thoroughly explored the PESs of DMSO clusters from dimer to decamer and located up to 159 configurations. We used a dispersion-corrected DFT functional, PW6B95-D3, benchmarked to be among the accurate functionals to study DMSO clusters. This functional allowed us to calculate accurate binding electronic energies of DMSO clusters.



Comments (0)